| |
|||||||||||||||||
| Série de monographies sur les maladies liées au vieillissement : XII. Maladie de Parkinson - Percées récentes et nouvelles orientations Résumé La maladie de Parkinson est une affection chronique progressive du système
nerveux central, caractérisée par un tremblement, une rigidité
et une bradykinésie, qui touche principalement les personnes de
plus de 50 ans. Les dernières recherches sur la maladie de Parkinson
se sont notamment attachées à évaluer le rôle
éventuel de l'alimentation et ont mis l'accent sur la génétique
et les facteurs héréditaires. L'identification de marqueurs
biologiques et d'autres facteurs de risque environnementaux tiendra une
large place dans la recherche future sur la maladie, car elle pourrait
guider l'élaboration des stratégies de prévention.
La maladie de Parkinson (MP) est une affection neurodégénérative qui se manifeste principalement par une atteinte de la coordination et de la motricité volontaire. C'est une maladie qui s'observe dans la deuxième moitié de la vie, généralement après l'âge de 50 ans. Bien que la découverte de la MP soit généralement attribuée à James Parkinson, qui l'a décrite en 1817 dans sa monographie intitulée The Shaking Palsy1 (la paralysie agitante), on trouve des descriptions de syndromes parkinsoniens dans les anciens textes ayurvédiques de l'Inde, datant de 4500 à 1000 av. J.-C.2. La première percée importante dans la recherche sur la MP a eu lieu dans les années 60, lorsqu'on a émis l'hypothèse du déficit en dopamine et introduit le traitement à la lévodopa1. Depuis, des progrès considérables ont été accomplis dans la définition, le diagnostic et la surveillance de la maladie, ainsi que dans la connaissance de l'étiologie, de l'évolution, et du traitement de la MP. On ne connaît pas encore la cause de la maladie de Parkinson et on est impuissant à la guérir, mais les recherches menées au cours des dernières années nous ont permis d'acquérir une meilleure compréhension de la question. Le présent document fait un survol des connaissances actuelles sur la MP et insiste tout particulièrement sur les percées récentes et les orientations futures de la recherche dans ce domaine.
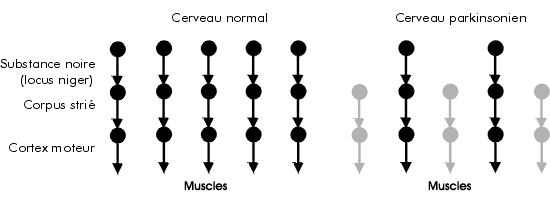
La MP est une affection chronique progressive du système nerveux central. C'est le plus courant des syndromes parkinsoniens, terme regroupant un ensemble de troubles de l'appareil moteur essentiellement caractérisés par trois symptômes : tremblement, rigidité et bradykinésie. Selon la majorité des études, le diagnostic de parkinsonisme ne peut être posé qu'en présence d'au moins deux de ces trois signes3-5. Les syndromes parkinsoniens se manifestent lorsque les neurones qui constituent la substantia nigra («substance noire» ou locus niger de Soemmering) sont détruits4. Le neurotransmetteur appelé dopamine est sécrété par les neurones du locus niger. Ces neurones sont reliés à d'autres neurones présents dans le corps strié (striatum) qui, à leur tour, transmettent les messages aux zones de commande du mouvement du cortex. La raréfaction neuronale dans le locus niger se traduit par un déficit en dopamine; on observe donc une diminution des signaux transmis en direction du corps strié et, de là, en direction du cortex. Le fonctionnement normal de l'appareil moteur s'en trouve donc perturbé (figure 1). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
La MP se distingue de la plupart des autres formes de parkinsonisme par le fait qu'on ignore la cause de la destruction de la substance noire conduisant au déficit en dopamine4. De nombreuses hypothèses ont été avancées, notamment celle du «stress oxydatif»6,7, pour expliquer l'apparition de la MP, mais aucune ne fait entièrement l'unanimité. La classification pathologique de la MP est notamment fondée sur la dégénérescence de certains groupes précis de cellules nerveuses, dont celles de la substance noire4. Ni les troubles circulatoires, ni l'artériosclérose ne permettent d'expliquer l'emplacement des cellules atteintes. Le diagnostic de MP peut également être posé après le décès à l'observation des corps de Lewy, inclusions de forme arrondie présentes dans les neurones atteints. Les corps de Lewy sont extrêmement évocateurs de la maladie.
Morbidité Selon un récent Rapport sur la santé dans le monde, on dénombre 3 765 000 personnes atteintes de MP dans le monde entier, et la maladie est diagnostiquée chez 305 000 personnes chaque année8. En 1996, on a recensé 2 635 000 personnes atteintes de MP souffrant d'incapacité chronique et 58 000 décès. Bien que la MP soit présente à l'échelle planétaire, indépendamment de l'appartenance ethnique et de la situation socio- économique, les statistiques relatives à la morbidité et à la mortalité varient considérablement d'un endroit à l'autre. En fait, une récente analyse de la distribution de la MP dans le monde9 a mis en évidence une différence de 13 ordres de grandeur entre les estimations les plus élevées (Uruguay) et les plus faibles (Chine) de la prévalence dans des études de porte-à-porte, et une différence de trois ordres de grandeur entre les endroits affichant la plus forte (Islande) et la plus faible (Libye) prévalence dans des études fondées sur des données provenant de sources telles que les hôpitaux, les médecins et les dossiers d'assurance-maladie. Dans les estimations de l'incidence, on observait une différence de 10 ordres de grandeur entre les régions ayant les taux les plus élevées (États-Unis) et les plus faibles (Chine). Ces écarts dans la prévalence et l'incidence donnent à penser que des facteurs environnementaux ou génétiques pourraient jouer un rôle dans la genèse de la maladie. Toutefois, d'autres facteurs pourraient également expliquer ces écarts, comme des méthodes diagnostiques ou des groupes de population différents. Étant donné que dans cette analyse les taux de toutes les études ont été standardisés pour une seule population, les variations ne peuvent être attribuées aux différentes structures d'âge des populations. On peut avoir recours à la détermination des cas pour expliquer l'écart dans les estimations entre les études de porte-à-porte et les autres études qui ne recherchent pas activement les personnes atteintes de MP. Dans le passé, la littérature a signalé de façon relativement constante des taux plus élevés de MP dans les populations majoritairement de race blanche que dans les populations asiatiques ou noires. Des études plus récentes indiquent que la variation de la prévalence de la MP entre les différents groupes ethniques est moins importante qu'on ne l'avait déjà cru, mais la prévalence demeure variable d'une étude à l'autrel0-17. Ce changement pourrait être attribuable à l'uniformité accrue des méthodes d'étude. Les études récentes de l'incidence menées aux États-Unis et en Europe ont unanimement fait ressortir des taux d'incidence de la MP s'échelonnant entre 8 et 13 pour 100 000 habitants15,18-20. Toutes les études comportant des données selon le sexe ont fait état de taux d'incidence plus élevés chez les hommes que chez les femmes. Le taux d'incidence élevé observé chez les hommes dans une étude effectuée à Manhattan (États-Unis)15 est en grande partie imputable au taux d'incidence chez les hommes de race noire; ce dernier était systématiquement plus élevé que chez les hommes de race blanche, dans tous les groupes d'âge, et l'on observait une différence de quatre ordres de grandeur chez les sujets de plus de 80 ans. La maladie est probablement de plus courte durée chez les hommes noirs, car la même étude a fait état de plus faibles taux de prévalence chez les hommes noirs que chez les hommes blancs. Les auteurs d'une étude effectuée en Suède ont utilisé les dossiers d'un organisme d'assurance-maladie (OAM)20 et observé un taux brut d'incidence plus élevé chez les Blancs que chez les Noirs, les Hispaniques ou les Asiatiques; ce sont les Asiatiques qui avaient le plus faible taux. Dans une cohorte de résidents hawaïens de sexe masculin, dont les ancêtres étaient principalement originaires du Japon et d'Okinawa18, le taux d'incidence se situait à mi-chemin entre les taux de l'étude de Manhattan et ceux de l'étude de l'OAM. Les auteurs ont conclu que les facteurs environnementaux jouent un rôle plus important que les facteurs génétiques dans ce groupe d'hommes, étant donné que les taux d'incidence signalés antérieurement chez les Asiatiques étaient plus bas. Toutes les études ont fait ressortir des taux d'incidence qui augmentaient de façon linéaire jusqu'à l'âge de 75 ans. À cet âge, dans la majorité des groupes, le taux d'incidence plafonnait ou continuait d'augmenter de façon linéaire. Dans la cohorte hawaïenne, toutefois, le taux d'incidence diminuait, alors qu'il augmentait de façon encore plus marquée chez les hommes de Manhattan. Morbidité au Canada Rares sont les données faisant état de la prévalence et de l'incidence de la MP au Canada. Étant donné que la majorité des patients n'ont pas besoin d'être hospitalisés, les taux de congés des hôpitaux conduisent à une sous-estimation de la prévalence de la MP. Ces taux présentent en outre l'inconvénient de n'être pas fondés sur le nombre de sujets mais plutôt sur le nombre de congés. Il est donc possible qu'un même sujet soit compté plus d'une fois. En dépit de ces faiblesses, les taux de congés des hôpitaux peuvent aider à déceler les tendances et les différences générales. Le taux global de congés des hôpitaux pour la MP (code de la maladie de Parkinson, 332.0, CIM-9) entre 1991 et 1995 était de 15,4 pour 100 000 chez les hommes et de 8,9 pour 100 000 chez les femmes (tableau 1). On observait des variations considérables d'une province à l'autre, le taux s'échelonnant entre 8,0 pour 100 000 à Terre-Neuve et 19,3 pour 100 000 en Saskatchewan. Cette variation s'explique en partie par l'utilisation de codes de maladies ou de critères d'hospitalisation différents. Entre 1991 et 1995, les taux de congés des hôpitaux augmentaient avec l'âge, pour atteindre un sommet chez les personnes âgées de 80 à 84 ans. Dans tous les groupes d'âge, les taux d'hospitalisation étaient plus élevés chez les hommes que chez les femmes (tableau 2), et les taux globaux de congés des hôpitaux étaient plus élevés chez les hommes dans toutes les provinces. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Au cours de la période 1976-1980 à 1991-1995, les taux de congés des hôpitaux liés à la MP ont chuté de 25 % dans tous les groupes d'âge. Les taux ont diminué chez les femmes à partir de 1976-1980, mais ils ont atteint un sommet chez les hommes au cours de la période 1986-1990. La diminution des taux de congés des hôpitaux pourrait être attribuée au déclin enregistré dans les groupes d'âge plus jeunes, en particulier chez les femmes. Dans les tranches d'âge supérieures, les taux de congés des hôpitaux ont en fait augmenté. Si l'on exclut les données enregistrées de façon systématique, comme les taux de congés des hôpitaux, il y a eu très peu de tentatives documentées d'estimer la prévalence de la MP au Canada. Une étude menée en 1988 dans une collectivité rurale de la Colombie-Britannique21 a mis en évidence un taux brut de prévalence de 69 pour 100 000, ce qui est jugé faible comparativement à celui des autres collectivités. On n'a toutefois pas précisé les taux selon l'âge, aussi les comparaisons sont-elles impossibles. Une autre étude a été menée auprès d'une cohorte de personnes couvertes par le régime d'assurance-maladie de l'Alberta, qui ont fait l'objet d'un suivi de cinq ans22. Les patients parkinsoniens ont été repérés à partir des données sur la facturation des médecins dans lesquelles figuraient un code diagnostique de MP. Le taux brut de prévalence s'élevait à 244,4 pour 100 000. Le taux était plus élevé chez les hommes que chez les femmes, et 81 % des sujets atteints avaient 60 ans et plus. Les auteurs d'une étude effectuée en Saskatchewan ont observé que 3 % des sujets de plus de 65 ans souffraient de la MP23. Cette estimation est relativement instable, étant donné qu'elle n'était fondée que sur les deux seuls cas positifs parmi les 70 sujets. Une étude analogue a mis en évidence un taux de 6 % de MP dans un établissement de soins prolongés24. Des études menées dans d'autres pays ont révélé que la prévalence plus élevée de la MP chez les sujets vivant dans un établissement de soins prolongés est en grande partie imputable à la plus forte prévalence de la maladie chez le groupe d'âge «jeune-vieux»25,26.
Les taux de mortalité à l'échelle internationale augmentent avec l'âge et sont constamment plus élevés chez les hommes. Selon les données publiées récemment, les taux de mortalité sont semblables dans les pays européens27-29 et plus bas au Japon30. Les taux de mortalité ont augmenté de façon soutenue dans les populations plus âgées (>75 ans), et diminué dans les populations plus jeunes (<65 ans)31.
Bien que la plupart des patients atteints de MP ne meurent pas de cette maladie, les données sur la mortalité permettent de repérer les différences dans la distribution de la maladie selon la région géographique, le sexe, l'âge et le temps. Les taux de mortalité peuvent en outre faire ressortir les différences dans le traitement et la prise en charge. Entre 1992 et 1996, le taux global de mortalité due à la MP était de 3,4 pour 100 000 habitants (tableau 3). Dans deux provinces, les taux de mortalité s'écartaient de façon significative du taux national : l'Ontario affichait un taux significativement plus élevé (3,7 pour 100 000), et l'Alberta un taux significativement plus bas (2,6 pour 100 000) pour tous les âges. Comme dans le cas des autres taux, ces différences peuvent être réelles ou être imputables à d'autres facteurs, comme les différences interprovinciales dans les codes indiqués sur les certificats de décès. Entre 1992 et 1996, les taux de mortalité ont augmenté avec l'âge et n'ont pas atteint un sommet comme les taux de congés des hôpitaux (tableau 4). Dans tous les groupes d'âge, les taux de mortalité étaient plus élevés chez les hommes que chez les femmes et, à l'instar des taux de congés des hôpitaux, les taux globaux de mortalité étaient plus élevés chez les hommes dans toutes les provinces. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Au fil du temps, les taux de mortalité standardisés ont augmenté aussi bien chez les hommes que chez les femmes. L'augmentation observée chez les hommes entre 1977-1981 et 1992-1996 était plus importante (93 %) que chez les femmes (79 %). Comme dans le cas des taux de congés des hôpitaux, l'augmentation de la mortalité due à la MP est en grande partie due à une plus forte hausse de la MP dans les groupes plus âgés que dans les groupes plus jeunes. Les taux de mortalité dans les tranches d'âge inférieures n'ont cependant pas diminué dans la même mesure que les taux de congés des hôpitaux.
Facteurs environnementaux Des efforts considérables ont été consacrés à la recherche d'un agent environnemental responsable de la MP. Cette recherche a été particulièrement intensive entre le milieu et la fin des années 80, lorsqu'on a découvert que le MPTP (1-méthyl-4-phényl-1,2,3,6-tétrahydropyridine), un contaminant rare de l'héroïne, entraînait un syndrome à peu près identique, aux plans clinique et histologique, à la MP32,33. On a alors cru qu'une toxine présente dans l'environnement, ayant des propriétés chimiques et physiques semblables à celles du MPTP, pouvait entraîner la MP. Bien qu'aucune toxine n'ait pu être mise en cause dans la genèse de la MP, les études ont permis de recueillir des données suffisantes pour éliminer certaines hypothèses et en examiner d'autres plus à fond.
Si, dans le passé, certaines études ont fait ressortir un lien entre l'habitat en milieu rural et la MP, les dernières études ont donné des résultats assez contradictoires. Ce manque de consistance caractérise non seulement les résultats, mais aussi les périodes d'exposition à l'étude. Deux études ont mis en évidence des rapports de cotes (RC) élevés et significatifs pour l'habitat en milieu rural peu de temps avant le diagnostic34,35, alors qu'une autre étude a établi que la mortalité due à la MP était positivement liée à la densité de la population36. Les auteurs d'une étude effectuée en Chine ont observé un RC inférieur à 1 pour l'habitat dans des petites villes, mais ils n'ont pas précisé la période d'exposition à l'étude37. Une étude américaine a mis en évidence un RC élevé et significatif pour des antécédents d'habitat en milieu rural chez les Noirs seulement, mais non dans les autres groupes ethniques38. Dans une étude, on n'a observé aucune différence entre les cas et les sujets témoins en ce qui concerne la densité moyenne de population des lieux de résidence entre la naissance et le moment du diagnostic39; une autre étude n'a révélé aucun lien entre le lieu de résidence pendant les 15 premières années de vie et le risque de MP40. Comme on croit que l'apparition de la MP est précédée d'une longue période de latence asymptomatique41, la période d'exposition probable présentant un intérêt serait située au début de la vie plutôt qu'à un âge plus avancé. Vu que de nombreux types particuliers d'exposition sont associés aux régions rurales, des études récentes ont tenté de mesurer ces expositions afin de tenter d'expliquer le lien observé entre la MP et les régions à faible densité de population. Trois études récentes36,42,43 ont mis en évidence un lien entre la MP et le travail agricole. L'une d'elles43 était exclusivement fondée sur les certificats de décès, et une autre36 a observé un lien positif entre le nombre de décès dus à la MP et la densité agricole. Dans la troisième étude42, le RC pour la production de grains et de grandes cultures était significatif dans une analyse unidimensionnelle, mais non dans une analyse multidimensionnelle. Une étude allemande a fait ressortir un risque relatif élevé et significatif associé à la cueillette des champignons pendant l'enfance et l'adolescence44; aucun lien n'a toutefois été observé avec l'activité agricole antérieure ou le travail en milieu agricole, le fait d'habiter une ferme ou d'habiter à proximité d'une ferme, d'avoir des contacts avec des animaux d'élevage ou d'être associé à l'abattage. D'autres études récentes34,38 n'ont pas montré de lien entre le risque de MP et les activités agricoles.
La similitude observée entre la structure du MPP+, métabolite du MPTP, et celle d'un herbicide, le paraquat, a encouragé la recherche d'un lien entre l'exposition aux pesticides et la MP (tableau 5)34,35,39,45-48. Une étude effectuée à Taïwan a indiqué un RC de 2,0 pour la MP chez les sujets qui avaient utilisé à la fois le paraquat et d'autres herbicides ou pesticides, comparativement à ceux qui avaient été exposés à des pesticides et herbicides autres que le paraquat48. Les dernières études ont montré de façon constante un risque accru de MP associé à l'exposition aux pesticides et, dans certaines études, ces résultats atteignaient le niveau de signification statistique. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Eau de puits L'eau de puits a également été mise en cause dans la MP. Parmi les études récentes, seules celles qui ont été menées en Italie49 et en Espagne50 ont fait ressortir un lien positif entre la consommation d'eau de puits et le risque de MP. Une étude qui s'est intéressée au lien éventuel entre la mortalité due à la MP et la proportion de consommateurs d'eau de puits au Michigan a mis en évidence un lien négatif entre l'exposition et la maladie36; une étude effectuée en Chine a également signalé un risque diminué de MP associé à la consommation d'eau de puits37. Quatre études n'ont découvert aucun lien38-40,48, et on n'a trouvé dans l'eau de puits aucun contaminant considéré comme un facteur étiologique possible de la MP.
On a découvert que certains métaux, comme le manganèse et le mercure, pouvaient induire des signes et des symptômes parkinsoniens chez des sujets ayant subi une exposition aiguë importante51. Les auteurs d'études épidémiologiques récentes se sont principalement intéressés à l'exposition professionnelle aux métaux en tant que facteur de risque de MP. Aucun RC significatif n'a été mis en évidence dans les études effectuées en Colombie-Britannique47 ou en Alberta45. Une étude allemande n'a trouvé qu'un seul RC significatif pour l'exposition professionnelle au plomb39. Toutefois, ce dernier était à peine significatif, et ne l'était qu'en comparaison avec l'un des deux groupes témoins. On a observé un autre RC qui était tout juste significatif par rapport à un seul groupe témoin pour l'exposition au mercure due aux amalgames dentaires. Selon une étude écologique, les comtés du Michigan où l'on retrouvait des industries métallurgiques (fer et cuivre) affichaient des taux de mortalité due à la MP plus élevés36. Dans une autre étude, les RC élevés observés pour une exposition professionnelle de 20 ans au cuivre et au manganèse et à diverses combinaisons de métaux donnent à penser que l'exposition aux métaux pourrait jouer un rôle dans la genèse de la MP52. Les auteurs d'une autre étude ont cependant découvert que les taux de manganèse, de chrome et de cobalt dans le sérum et l'urine des patients parkinsoniens ne différaient pas de ceux des témoins, ce qui les a amenés à conclure à l'absence de lien entre ces métaux et la MP53.
On a également examiné la relation éventuelle entre la MP et de nombreuses autres toxines. Bien que les auteurs d'une étude54 aient fait état de liens positifs entre les résines plastiques, les résines époxides, les colles, les peintures et le pétrole, ils ont également conclu qu'une multitude d'autres expositions n'étaient pas significatives, ce qui rendait difficiles les comparaisons multiples. Des taux plus élevés de mortalité attribuable à la MP ont été observés dans les pays ayant des industries papetières et chimiques36. D'autres études n'ont observé aucune relation avec les toxines industrielles35, le monoxyde de carbone39,45, le cyanure45, les fumées d'échappement39, ni avec les colles, les peintures et les vernis-laques39. Les patients parkinsoniens étaient plus nombreux que les patients témoins à signaler la présence dans leur maison de murs recouverts de panneaux de bois55; cette observation pourrait être en faveur d'un lien étiologique entre les produits de préservation du bois et la MP.
Les traumatismes crâniens ont été mis en cause dans la genèse de la MP, probablement en raison des cellules microgliales associées au processus inflammatoire6. Deux études récentes sur quatre35,39,45,56 ont observé des RC significatifs pour le traumatisme crânien. Dans l'une de ces études, le RC était à peine significatif et ne l'était qu'en comparaison avec l'un des groupes témoins39; l'autre étude faisait état d'un RC qui demeurait significatif après une analyse multidimensionnelle45. Les études portant sur le traumatisme crânien pourraient comporter un biais de mémoire, aspect dont devraient tenir compte les études futures.
Les études épidémiologiques ont constamment montré que le tabagisme avait un effet protecteur à l'égard de la MP. La majorité des études récentes semblent corroborer cette relation, car elles ont fait état de RC inférieurs à 1 (tableau 6)34,39,40,45,56-59. Outre ces données, une étude prospective menée dans le cadre de l'Honolulu Heart Study a fait ressortir un risque relatif significatif de 0,3960. Certaines données expérimentales confirment l'hypothèse voulant que la nicotine ait un effet protecteur à l'égard de la MP. Une étude a montré que la consommation chronique de nicotine chez les rats ralentissait la diminution des récepteurs dopaminergiques et du recaptage de la dopamine associée au vieillissement61.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
D'autres études cas-témoins ne corroborent toutefois pas l'effet protecteur du tabagisme à l'égard de la MP. L'analyse longitudinale de Gompertz, qui prend en compte les trois dimensions de la génétique, de l'environnement et de la mortalité précoce sélective, montre que l'hypothèse d'un effet neuroprotecteur n'explique pas le lien négatif entre la MP et le tabagisme62. Ce lien négatif a été attribué au fait que les fumeurs meurent plus jeunes que les non-fumeurs. Tzourio et coll.58 n'ont observé aucun effet protecteur du tabagisme à l'égard de la MP, mais une fois les données corrigées pour tenir compte de l'âge, on a observé que le tabac avait un effet protecteur chez les sujets plus jeunes, alors qu'il contribuait à accroître le risque chez les sujets plus âgés. Bien que le tabagisme ait un effet protecteur à l'égard de la MP, il demeure un important facteur de risque pour de nombreuses autres maladies graves, et ses effets indésirables dépassent largement ses bienfaits potentiels.
Le régime alimentaire vient tout juste d'être mis en cause dans l'étiologie de la MP. Selon l'hypothèse du stress oxydatif, une augmentation des antioxydants devrait contribuer à prévenir la dégénérescence et la mort des neurones dopaminergiques par l'élimination d'un plus grand nombre de radicaux libres. Par conséquent, les antioxydants fournis par les aliments et les suppléments auraient un effet protecteur à l'égard de la MP. Les études épidémiologiques qui ont examiné le lien éventuel entre les antioxydants et la MP ont donné des résultats contradictoires (tableau 7)63-68; jamais la mise en évidence d'un lien négatif ou positif n'a pu être répétée dans une autre étude. Les deux études prospectives63,65 ont utilisé les données relatives aux antécédents alimentaires recueillies avant le diagnostic de MP; dans une étude63, il ne s'était écoulé que quelques années entre la collecte des données sur l'alimentation et l'apparition de la maladie63. Étant donné que le processus morbide serait, croit-on, amorcé de nombreuses années avant que le sujet ne devienne symptomatique, les données relatives à l'alimentation obtenues dans la dernière étude pourraient n'être d'aucune utilité dans la détermination de l'étiologie de la maladie. Les trois autres études64,66,67 n'ont guère fait mieux, car il s'agissait d'études cas-témoins dont les données étaient axées sur les habitudes alimentaires au cours de l'année précédente. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Trois autres études cas-témoins34,44,68 ont examiné la relation éventuelle entre la MP et les aliments riches en vitamine E. Deux de ces études44,68 n'ont pas fait ressortir de différence dans la consommation d'aliments riches en vitamine E, mais la troisième a fait état d'un RC élevé et significatif pour les noix et les graines, qui sont des aliments riches en vitamine E34. L'une de ces études68 a mis en évidence un apport plus élevé en vitamine C chez les patients atteints de MP. Certaines études ont également examiné le lien éventuel entre les concentrations sériques d'antioxydants et la MP. Trois études69-71 n'ont pas observé de différence entre les patients parkinsoniens et des sujets témoins en bonne santé en ce qui concerne les concentrations sériques de vitamine E. L'une de ces études70 n'a pas non plus observé de différence entre les concentrations de vitamine A, mais a signalé de plus fortes concentrations de vitamine C chez les patients parkinsoniens; chez les sujets témoins, les concentrations de vitamine C étaient cependant faibles comparativement aux données établies chez des sujets jeunes en bonne santé. Il importe de remarquer que ces études étaient fondées sur les concentrations mesurées après le moment du diagnostic et pourraient ne pas refléter les concentrations antérieures à l'apparition de la maladie. On a aussi examiné récemment le lien éventuel entre d'autres variables liées au régime alimentaire et l'étiologie de la MP. Une étude écologique a fait état de liens significatifs et positifs entre les taux de mortalité ajustés pour tenir compte de l'âge dans 17 pays différents et la consommation de protéines alimentaires totales et de viande par habitant72. En plus de présenter les faiblesses inhérentes aux études écologiques, cette étude utilisait non seulement les taux de mortalité, qui, contrairement aux autres données statistiques, n'indiquent pas avec précision la prévalence de la MP, mais également les chiffres de 1952 à 1958, qui ne reflètent pas les taux actuels. Deux études cas-témoins64,66 ont fait ressortir un RC élevé et significatif pour l'apport en matières grasses. L'une de ces études66 a également signalé des RC élevés et significatifs pour le cholestérol, le fer et la lutéine. Vu que les lipides constituent l'une des principales sources de radicaux libres, l'augmentation de l'apport en matières grasses et en cholestérol est compatible avec l'hypothèse du stress oxydatif. Le lien positif observé avec la lutéine pourrait être imputable à la MP, car de nombreux patients augmentent leur consommation de lutéine pour atténuer les symptômes de la maladie. Selon une étude allemande, la MP pourrait être liée à une gamme d'aliments73. Les patients parkinsoniens consomment davantage de chocolat, de desserts, d'abats et de viandes crues, et moins de bière et de café. Le lien entre la maladie et ces denrées alimentaires pourrait être attribuable aux effets des amines biogènes (chocolat), des taux d'insuline (aliments riches en glucides raffinés), des agents infectieux (abats et viandes crues), de l'éthanol (bière) et de la caféine (café) sur le système dopaminergique. Une autre étude a également signalé un lien négatif entre la consommation d'alcool et la MP74.
L'hypothèse de l'origine infectieuse de la MP s'explique en grande partie par l'apparition de symptômes parkinsoniens chez des sujets infectés par le virus responsable de l'encéphalite léthargique qui a sévi dans les années 2075. De nombreuses études se sont avérées impuissantes à mettre en évidence un lien entre la MP et une gamme de bactéries et de virus courants. Aucune étude récente n'a découvert de relation entre la MP et la varicelle, la rougeole, la rubéole, les oreillons, la grippe espagnole45 et les espèces du genre Nocardia76. Selon une étude effectuée au Royaume-Uni, les patients parkinsoniens étaient plus nombreux à se rappeler avoir souffert du croup ou de la diphtérie pendant l'enfance56. Il importe toutefois de noter que ces résultats ne sont pas fondés sur les taux d'anticorps sériques et que la neurotoxine produite par le micro-organisme responsable de la diphtérie ne peut franchir la barrière hémato-encéphalique. Une autre hypothèse étiologique mettant en cause la coqueluche a été mise de l'avant lorsqu'on a observé, en Islande, un lien positif entre la MP et les épidémies de coqueluche dans des cohortes de naissance d'une année77. Les patients parkinsoniens avaient également une réaction immunitaire plus forte aux coronavirus que des sujets témoins en bonne santé, ce qui est en faveur d'un lien entre les virus à ARN et la MP78. L'observation selon laquelle les ulcères gastro-intestinaux seraient plus répandus chez les patients parkinsoniens a amené à s'interroger sur le rôle éventuel d'Helicobacter pilori, bactérie à Gram négatif responsable de la majorité des cas d'ulcères79.
Pendant de nombreuses années, les épidémiologistes se sont surtout intéressés aux facteurs de risque environnementaux. L'existence d'une composante héréditaire semblait moins probable, car les études réalisées sur des jumeaux dans le passé avaient montré des taux de concordance semblables chez les jumeaux monozygotes et dizygotes55,80,81. Toutefois, plus récemment, les facteurs génétiques ont suscité un regain d'intérêt, en grande partie attribuable à la découverte de l'importance des antécédents familiaux en tant que facteur de risque de MP.
De nombreuses études épidémiologiques ont examiné le lien éventuel entre le risque de MP et des antécédents familiaux de maladie (tableau 8)34,35,37,39,45,49,82-85. La majorité, sinon la totalité, de ces études, ont signalé de façon constante un RC significativement élevé pour des antécédents familiaux. Il se peut que les bais de mémoire et de sélection expliquent dans une certaine mesure le lien observé, mais la cohérence, la force et l'universalité des résultats sont en faveur d'une influence des expositions environnementales à un âge précoce ou d'une certaine prédisposition génétique sous-jacente à la maladie. Une étude de Uitti et coll.86 a en outre permis de repérer des cas jusque-là non diagnostiqués de MP dans des familles qui n'avaient pas signalé d'antécédents de maladie, ce qui met en doute l'exactitude des renseignements fournis par les patients à cet égard. Les résultats de cette étude indiquent en outre que le taux pondéré de prévalence du parkinsonisme familial est plus de 5 fois supérieur aux taux déclarés de prévalence de la MP dans l'ensemble de la population. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
La littérature a signalé l'existence d'un certain nombre de familles où les cas de MP étaient nombreux. L'un des exemples les plus frappants est celui d'une famille comptant 18 personnes atteintes sur six générations87. Les sujets présentaient les anomalies caractéristiques de la MP à l'autopsie; en outre, les symptômes cliniques, notamment l'âge de début et la réponse à la lévodopa, étaient ceux d'un cas classique. La famille Contursi, comptant 60 sujets parkinsoniens sur cinq générations, est un autre exemple intéressant88. Dans une famille gréco-américaine dont 16 membres étaient atteints sur trois générations, on observait une rigidité asymétrique, un tremblement de repos, une bradykinésie et une instabilité posturale qui répondaient également à la lévodopa89. Les données relatives à toutes ces familles étaient compatibles avec une transmission autosomique dominante à faible pénétrance. Une comparaison entre des cas familiaux et sporadiques de MP a en outre révélé que les paramètres cliniques et l'évolution de la maladie étaient semblables90.
Marqueurs génétiques Bon nombre de chercheurs estiment que l'identification du gène dans une famille constitue la percée la plus importante à avoir été effectuée dans la recherche sur la MP depuis la mise en évidence du déficit en dopamine91 et l'introduction ultérieure de la lévodopa comme traitement symptomatique92. Un article a d'abord signalé que des marqueurs génétiques présents sur le chromosome 4q21-q23 étaient liés aux sujets parkinsoniens dans une grande famille italienne93. Moins d'un an plus tard, un deuxième article décrivait précisément le gène et la mutation que l'on croyait responsables de la MP dans cette famille et dans d'autres familles (familles grecques)94. On a observé la substitution d'une paire de bases dans le gène de l'alpha-synucléine chez les membres atteints de ces familles, caractéristique qui était absente chez les sujets non atteints et chez des cas sporadiques de MP. On ne connaît pas la fonction de la protéine codée par ce gène; on a toutefois émis l'hypothèse que la forme mutée s'agglutine avec ses semblables dans les terminaisons nerveuses, entraînant la mort neuronale. Cette mutation, croit-on, n'expliquerait qu'une petite fraction des cas familiaux de MP, mais on espère que cette découverte permettra d'élucider d'autres cas de MP. De nombreux autres gènes ont fait l'objet d'études visant à établir un lien entre des facteurs héréditaires et la MP. La famille d'enzymes cytochrome P-450 est responsable de la détoxification de nombreux médicaments et agents environnementaux95. En raison du polymorphisme de la débrisoquine hydroxylase (CYP 2D6), l'intensité du métabolisme varie d'une personne à l'autre. S'il est vrai que les expositions aux agents environnementaux jouent un rôle dans la MP, on suppose que le risque de MP pourrait être accru par l'insuffisance de la détoxification de ces agents. On observerait alors une augmentation de la quantité de toxines pouvant intervenir en divers points du processus de stress oxydatif. Les auteurs d'études antérieures ont examiné les phénotypes des sujets en leur administrant de la débrisoquine par voie orale puis en dosant le métabolite dans les urines. Les sujets ont été divisés en bons et piètres «métaboliseurs», en fonction du pourcentage de débrisoquine récupéré dans l'urine. Étant donné qu'aucune étude menée chez des sujets de race blanche n'avait mis en évidence un RC significatif96-102 chez les piètres métaboliseurs, on a effectué des études axées sur le génotype des sujets. Ces dernières études faisaient notamment appel à l'analyse directe du gène CYP 2D6 et à la détermination du variant spécifique associé au phénotype du piètre métaboliseur. La divergence entre les études était très grande, tant au chapitre des résultats que du nombre de variants inclus dans l'analyse génétique. Les résultats concernant le lien éventuel entre le risque de MP et le variant le plus courant, CYP 2D6B103-112, n'ont pas permis d'établir de façon probante que le gène CYP 2D6 était lié à la MP, mais ce dernier pourrait néanmoins jouer un rôle dans un sous-groupe d'individus. D'autres membres de la famille cytochrome P-450, comme CYP 1A2 et CYP 3A4, pourraient aussi influer de façon importante sur la susceptibilité à la MP113,114. Un lien a été établi entre le génotype acétylateur lent de la N-acétyltransférase 2 et la MP familiale115. La susceptibilité du patient aux toxines environnementales pourrait s'en trouver accrue; il faudra toutefois mener d'autres recherches à ce sujet.
On a observé une baisse de l'activité du complexe I, groupe de protéines participant à la respiration aérobique, non seulement dans le tissu cérébral des patients parkinsoniens116-118, mais également dans les cellules hybrides119, le muscle squelettique120,121, les fibroblastes122 et, dans certaines études, les plaquettes123,124. On a également découvert que le complexe I était inhibé par le métabolite actif du MPTP125. Étant donné que sept des quelque 40 sous-unités du complexe I sont codées par l'ADN mitochondrial126 et que cet ADN est plus facilement endommagé que l'ADN nucléaire127, les altérations du génome mitochondrial, qu'elles soient héréditaires ou acquises par exposition à des agents toxiques, pourraient jouer un rôle central dans la neurodégénérescence associée à la MP. Les études ayant analysé les mutations de l'ADN mitochondrial ont initialement signalé une importante délétion du matériel génétique chez les patients parkinsoniens128. Des études ultérieures ont toutefois minimisé ces résultats et indiqué que cette délétion était un phénomène lié au vieillissement, indépendant de la MP129-131. D'autres anomalies des gènes mitochondriaux ont été observées dans le cerveau de patients parkinsoniens132-135, mais les plans expérimentaux de bon nombre de ces études ne comportaient pas de pondération pour tenir compte de l'âge. L'ADN mitochondrial étant exclusivement hérité de la mère, on pourrait s'attendre à observer une hérédité maternelle s'il était associé à la MP. Deux études136,137 qui se sont expressément intéressées à l'hérédité maternelle ont tenté de corroborer l'hypothèse selon laquelle la transmission d'un gène anormal serait responsable de la MP familiale; elles sont arrivées à des conclusions différentes. L'étude qui contredit cette hypothèse136 a simplement comparé le nombre de pères et de mères de patients parkinsoniens qui étaient également atteints de la maladie. Celle qui la corrobore137 n'a inclus que les familles dans lesquelles un parent et plusieurs membres de la fratrie étaient parkinsoniens. Selon les auteurs de la dernière étude, une simple analyse généalogique ne serait pas suffisamment sensible pour déceler la prépondérance de l'hérédité maternelle. Un phénomène d'anticipation génétique, caractérisé par une augmentation de la gravité de la maladie chez les générations ultérieures, a été signalé dans un certain nombre de familles ayant des antécédents de MP sur plusieurs générations89,138,139. Ce phénomène serait, croit-on, lié à l'expansion de répétitions de trinucléotides, comme dans le cas de maladies telles que la maladie de Huntington et la myotonie atrophique140. On n'a toutefois pas observé de différence dans l'expansion de répétitions de trinucléotides entre les patients parkinsoniens et les sujets témoins140,141, ni entre les générations dans les familles à MP où l'on observait un phénomène d'anticipation de l'âge de début140. L'analyse généalogique d'une grande famille indique que l'observation de l'anticipation pourrait être liée à une biais de constatation lié à l'âge88. Vu que, selon certains chercheurs, la MP présenterait des similitudes
avec la maladie d'Alzheimer (MA)142,143, le gène de
l'apolipoprotéine E (ApoE), qui est lié à la susceptibilité
à la MA144-147, a été le point de mire
d'autres études épidémiologiques génétiques.
Si l'on exclut une étude qui a signalé une plus grande fréquence
de l'allèle De nombreux autres paramètres génétiques et moléculaires ont été examinés récemment. Les études portant sur la superoxyde dismutase157, les récepteurs de la dopamine158 et la tyrosine hydroxylase159 ont donné des résultats négatifs, alors que des résultats positifs ont été obtenus dans celles portant sur les récepteurs de la lactoferrine160, la L-cystéine161,162, l'activité de la catalase163, le monoxyde d'azote163,164 et la catéchol-O-méthyl-transférase165. Les auteurs d'une étude ont examiné la liaison de nombreux gènes simultanément dans trois familles présentant un syndrome parkinsonien à transmission autosomique dominante166. Bien qu'ils aient obtenu, dans une famille, des résultats légèrement positifs pour le gène CYP 2D6, les données n'étaient pas en faveur d'une liaison des gènes pour la glutathion peroxydase, la tyrosine hydroxylase, le facteur neurotrophique d'origine cérébrale, la catalase, le précurseur amyloïde et la superoxyde dismutase à cuivre-zinc. Tout comme les facteurs environnementaux, les gènes qui jouent un rôle dans la pathogenèse de la MP peuvent être nombreux. Il est possible qu'une susceptibilité génétique limite la capacité du patient de détoxifier des facteurs environnementaux par ailleurs inoffensifs, ce qui entraînerait la dégénérescence des neurones dopaminergiques dans le système nigrostrié167.
Les monoamines oxydases (MAO) sont des enzymes de dégradation impliquées dans le métabolisme des toxines (types A et B)32,168-170 et dans la production de radicaux libres et de peroxyde d'hydrogène par la dégradation de la dopamine (type B)171-173. Tout comme les études génotypiques sur le CYP 2D6, les études portant sur les MAO ont donné des résultats très contradictoires. Certaines montrent un lien entre la MP et un polymorphisme du gène codant la MAO de type A (MAO-A) et non de type B, alors que d'autres mettent en évidence un lien entre la maladie et un polymorphisme de la MAO de type B (MAO-B) et non de type A (tableau 9)174-179. Les données semblent toutefois indiquer que la variabilité de l'enzyme MAO peut influer sur la pathogenèse et la progression de la MP. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
L'une des premières études examinant l'interaction gène-environnement dans la MP s'est intéressée au polymorphisme de la MAO-B et au tabagisme180. L'étude a fait ressortir un effet protecteur global du tabagisme à l'égard de la MP, analogue à celui observé dans les résultats exposés plus haut. Elle a toutefois également découvert que la relation inverse n'était présente que chez les sujets porteurs d'un certain variant de la MAO-B. Cette découverte permet non seulement d'ajouter une hypothèse génétique à la liste des idées avancées pour expliquer comment et pourquoi le tabagisme a un effet protecteur, mais elle souligne en outre l'importance des facteurs aussi bien génétiques qu'environnementaux dans l'étiologie de la MP.
La MP n'est pas toujours facile à diagnostiquer, étant donné qu'on ne peut se fier à un seul test diagnostique3,181. Au moins deux des trois principaux symptômes (tremblement, rigidité ou bradykinésie) doivent être présents pour que la maladie soit diagnostiquée. Il faut avoir exclu toutes les autres causes ou les autres formes de parkinsonisme. Une réponse positive aux médicaments dopaminergiques, comme la lévodopa, constitue également un critère diagnostique. On peut également décrire la MP sur le plan de la gravité de la maladie. La classification de Hoehn et Yahr permet d'exprimer le degré de handicap d'un sujet sur une échelle arbitraire comportant cinq stades. Le stade I est caractérisé par une atteinte unilatérale seulement, avec handicap habituellement minime ou nul; au stade V, le patient est grabataire ou ne peut, sans aide, se déplacer qu'en fauteuil roulant182. On a en outre signalé que plus du quart des patients parkinsoniens souffrent de démence et que certains patients atteints de la maladie d'Alzheimer présentent des signes de parkinsonisme183. Selon le Merck Manual of Geriatrics, il faut, pour poser le diagnostic, se fonder sur la présence des signes moteurs avant ou après le déclin cognitif183. La MP ne peut être diagnostiquée qu'après l'apparition des symptômes; au moment de l'extériorisation des premiers signes, environ 70 % des neurones de la substance noire ont déjà été détruits6. Cette observation suggère que la maladie évolue au cours d'une période asymptomatique pendant laquelle le sujet ne présente aucun signe clinique. Il serait donc utile de mettre au point une méthode de dépistage qui permettrait de repérer les sujets atteints au tout premier stade de la neurodégénérescence. L'intervention viserait alors à freiner le processus morbide, alors qu'aujourd'hui elle a principalement pour objet de traiter les symptômes. Bien qu'on ignore si la MP peut être décelée avant l'apparition des symptômes, de nombreuses stratégies ont été proposées184. Les études faisant appel à la tomographie par émission de positrons (PET), à la mesure du «temps de mouvement» et à la détermination des caractéristiques électrophysiologiques du tremblement montrent que ces méthodes peuvent aider à évaluer la dysfonction préclinique.
Bien qu'il soit impossible de guérir la MP, des traitements pharmacologiques et chirurgicaux sont disponibles. Le traitement de la MP est principalement pharmacologique et fait appel à différents médicaments destinés à accroître la quantité de dopamine présente dans le cerveau ou à réduire l'hyperactivité cholinergique (anticholinergiques)185-188. La dopamine ne peut traverser la barrière hémato-encéphalique, aussi est-il impossible d'administrer ce neurotransmetteur, c'est pourquoi on a introduit la lévodopa dans les années 60. La lévodopa est un précurseur de la dopamine, qui a longtemps été considérée comme la base du traitement de la MP; elle entraîne toutefois des effets indésirables qui consistent en des nausées et vomissements et une hypotension orthostatique. Bien qu'il soit largement admis que la lévodopa augmente les taux de survie, on ne s'entend pas sur le moment où il convient d'initier la thérapie189. La dopathérapie donne initialement de bons résultats, mais après plusieurs années, on observe chez la majorité des patients des fluctuations de réponse à la lévodopa (détérioration de fin de dose ou effet «on-off») ou des dyskinésies (mouvements anormaux involontaires)189. Les partisans d'une instauration précoce de la dopathérapie estiment que ces complications motrices reflètent l'évolution de la maladie, alors que les partisans d'un traitement plus tardif croient que la lévodopa pourrait avoir des effets toxiques. D'autres médicaments, comme les agonistes dopaminergiques, les anticholinergiques et l'amantadine, ont été introduits en association avec la lévodopa et ont principalement pour objet de réduire au minimum les effets indésirables de celle-ci. D'autres effets négatifs sont toutefois apparus. Depuis au moins une dizaine d'années, l'utilisation de la sélégiline, inhibiteur sélectif de la MOA-B, dans le traitement de la MP a été l'objet de controverse190-192. On n'a pas encore réussi à déterminer si ce médicament avait un effet neuroprotecteur à l'égard de la MP ou simplement un effet symptomatique. La présente série de monographies n'a pas pour objet d'examiner en détail les approches pharmacothérapeutiques; le lecteur qui s'intéresse à la question est prié de se reporter aux examens approfondis existants193,194. Outre la pharmacothérapie, le traitement de la MP fait appel à trois interventions chirurgicales195,196. Ce sont la chirurgie ablative, la stimulation cérébrale profonde et la greffe de tissu foetal. Les techniques de chirurgie ablative consistent à créer une lésion dans un circuit du pallidum (pallidotomie) ou du thalamus (thalamotomie). Étant donné que la dopamine module normalement un effet inhibiteur des noyaux gris centraux vers le thalamus, un déficit en dopamine se traduirait par une inhibition diminuée. Une lésion viendrait corriger la situation, car elle imiterait l'action de la dopamine en interrompant les signaux nerveux transmis entre le pallidum et le thalamus. La thalamotomie a donné de bons résultats chez des sujets souffrant de tremblements très invalidants. Bien que la pallidotomie permette d'atténuer la bradykinésie et la dyskinésie off, il faudra effectuer d'autres études pour évaluer les effets indésirables éventuels de la chirurgie197. La stimulation cérébrale profonde (SCP) ressemble à la chirurgie ablative, toutefois, plutôt que de créer une lésion, on implante une électrode de stimulation dans la cible. Selon une étude récente, la SCP et la thalamotomie permettent de soulager avec un égal succès le tremblement, mais la SCP serait préférable, car elle atténue les effets secondaires et contrôle la récurrence du tremblement sans autre chirurgie198. On a également effectué une greffe de tissu foetal chez certains patients parkinsoniens. Cette intervention consiste à implanter du tissu foetal sécrétant de la dopamine dans les noyaux gris centraux dans l'espoir que ce tissu se développe et produise en permanence de la dopamine chez le patient. En dépit des progrès, cette intervention demeure à un stade très expérimental et n'a encore été effectuée que chez un nombre limité de sujets, ayant fait l'objet d'un suivi de courte durée. Des activités de recherche et développement ainsi que des essais visant à trouver des médicaments plus efficaces tout en réduisant les effets indésirables sont en cours. Les symptômes parkinsoniens peuvent aussi être atténués dans une certaine mesure par une modification du régime alimentaire199 et des exercices précis200. Étant donné que les symptômes et la gravité de la maladie varient d'un patient parkinsonien à l'autre, il en est de même de la réponse à un même traitement. Les professionnels de la santé doivent donc collaborer étroitement avec leurs patients pour leur offrir le traitement correspondant le mieux à leurs besoins.
L'introduction de la lévodopa a permis d'accroître les taux de survie chez les patients parkinsoniens. Selon une étude récente201, l'utilisation de ce médicament a entraîné une amélioration marquée de la survie. Toutefois, ces bienfaits du traitement n'étaient observés que si la dopathérapie était instaurée aux tous premiers stades de la maladie. Bien que la dopathérapie et les autres types de traitement aient permis d'allonger l'espérance de vie, les patients parkinsoniens risquent encore davantage de mourir que des sujets du même âge. Une étude de cohorte menée en Angleterre et dans le pays de Galles auprès de patients parkinsoniens ayant fait l'objet d'un suivi de 20 ans a révélé que ces derniers risquaient deux fois plus de mourir que des sujets témoins de la population en général202, l'écart entre les hommes et les femmes étant très peu marqué. Une étude semblable menée en Écosse a mis en évidence un risque relatif de 2,5, mais la période de suivi n'était que de 3,5 ans203. Les auteurs de deux études américaines ont obtenu des risques relatifs de 2,7 avec un suivi moyen de 2,5 ans204, et de 2,0 avec un suivi moyen de 9,2 ans205. Dans une étude, un taux de mortalité plus élevé a été observé chez des sujets vivant en établissement et atteints de MP que chez des sujets ne souffrant pas de cette maladie206, mais une autre étude n'a pas donné le même résultat26. Dans l'une des études mentionnées, le risque de décès augmentait avec le nombre de signes parkinsoniens présents chez le malade205. La présence de troubles de la démarche, en particulier, était liée à un risque accru de mortalité. Il convient d'interpréter avec circonspection ces observations, car certaines de ces études intégraient des sujets souffrant de toutes les formes de parkinsonisme. Dans une étude portant exclusivement sur des sujets atteints de MP, la période écoulée entre le début de la maladie et les stades I, II, III, IV et V de la classification de Hoehn et Yahr était, respectivement, de 4,0, 6,5, 7,9, 9,8 et 11,8 ans207. Les patients qui avaient observé initialement des symptômes unilatéraux avaient un pronostic plus favorable que ceux dont les symptômes étaient d'emblée bilatéraux. Soixante-dix pour cent de l'ensemble des patients avaient observé initialement des symptômes unilatéraux, qui s'étaient généralisés dans 91 % des cas. Louis et coll. ont observé que la gravité des signes extrapyramidaux était l'unique et principal indicateur d'une mortalité accrue chez les patients parkinsoniens204. Les patients parkinsoniens diffèrent en outre de l'ensemble de la population en ce qui concerne la cause précise du décès. Selon certaines études, les patients parkinsoniens sont plus nombreux à mourir de cardiopathie ischémique202,207, d'accidents vasculaires cérébraux202,204,208, de pneumonie204,208,209 et d'autres affections respiratoires202. On s'explique mal ces différences, mais elles pourraient être attribuables à des causes concurrentes de décès, aux effets indésirables du traitement ou à une étiologie commune202. Certains auteurs ont observé une mortalité par cancer significativement plus faible chez des patients parkinsoniens que dans une population appariée selon l'âge et le sexe204,208. Cette observation ne s'applique toutefois qu'aux cancers que l'on croit liés au tabagisme et pourrait être attribuable au fait que les fumeurs sont moins nombreux chez les parkinsoniens210.
La dépression frappe davantage les sujets atteints de maladies physiques, tels que les accidents vasculaires cérébraux, le cancer et les troubles endocriniens ou métaboliques211. Les rares études récentes qui se sont intéressées au lien éventuel entre la dépression et la MP ont donné des résultats très contradictoires. Cette variabilité s'explique en grande partie par l'utilisation de critères différents pour mesurer la dépression. Toutefois, selon des études récentes, la dépression est plus répandue chez les patients parkinsoniens qui souffrent de démence que chez ceux qui n'en souffrent pas212-215. Chez les patients parkinsoniens, la dépression était également associée à des troubles de la pensée215 et à une dystonie neurovégétative216. Des incohérences ont également été observées en ce qui concerne le lien éventuel entre la dépression et le vieillissement, l'évolution de la maladie et la difficulté à accomplir les activités de la vie quotidienne214,215,217,218.
Conséquences pour une population vieillissante L'augmentation spectaculaire, au cours des trois prochaines décennies, de la proportion de la population canadienne et mondiale âgée de plus de 65 ans devrait s'accompagner d'une augmentation proportionnelle du nombre de sujets atteints de MP. Alors qu'en 1991, 11,6 % de la population canadienne était âgée de plus de 65 ans, ce groupe devrait représenter 23,6 % de la population d'ici 2016219. Cette hausse se traduira par une augmentation de 87 % du nombre de patients ayant besoin de soins médicaux en établissement, et une augmentation de 92 % du nombre de personnes qui mourront de la MP. Ce changement sera surtout manifeste dans les tranches d'âge supérieures, où l'on prévoit que le nombre de patients atteints de MP sera plus de deux fois plus élevé.
La recherche sur la MP a donné des résultats très intéressants au cours des dernières années, mais de nombreuses questions demeurent en suspens220. L'identification de marqueurs biologiques de la MP tiendra une place importante dans les recherches futures. Elle permettra aux chercheurs et aux médecins de diagnostiquer la maladie avec plus de précision, de mettre au point des traitements qui ralentiront ou arrêteront l'évolution de la maladie après son installation et de mettre en place des mesures de prévention. Elle permettra en outre de suivre de près les sujets courant un risque accru avant l'apparition des symptômes. La recherche génétique pourrait conduire à la découverte d'autres gènes qui prédisposent à la MP. L'examen plus approfondi du rôle éventuel des facteurs de risque environnementaux pourrait permettre d'obtenir des données précieuses qui guideront l'élaboration des stratégies de prévention. La compréhension de l'action réciproque des facteurs environnementaux et génétiques pourrait bien être la clé qui ouvrira la voie aux progrès futurs de la recherche sur la MP.
1. Duvoisin RC. History of parkinsonism. Pharm Ther 1987;32:1-17. 2. Manyam BV. Paralysis agitans and levodopa in "Ayurveda": ancient Indian medical treatise. Mov Disord 1990;5:47-8. 3. Pahwa R, Koller WC. Defining Parkinson's disease and parkinsonism. Dans: Ellenberg JH, Koller WC, Langston JW, réds. Etiology of Parkinson's disease. New York: Marcel Dekker Inc., 1995:1-54. 4. Duvoisin RC, Sage J. What is parkinsonism? Dans: Duvoisin RC, Sage J, réds. Parkinson's disease. A guide for patient and family. New York: Lippincott-Raven, 1996:1-13. 5. Rajput AH, Rozdilsky B, Rajput A. Accuracy of clinical diagnosis in Parkinsonism-a prospective study. Can J Neurol Sci 1991;18:275-8. 6. Youdim MBH, Riederer P. Understanding Parkinson's disease. Scientific American 1997;276:52-9. 7. Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology 1996;47:S161-70. 8. Organisation mondiale de la santé. Rapport sur la santé
dans le monde, 1997 : vaincre la souffrance, enrichir l'humanité.
Rapport du directeur-général. 9. Zhang ZQ, Hong X, Roman GC. Epidemiological characteristics of Parkinson's disease in various areas in the world [survol] (chinois). Chung-Hua Liu Hsing Ping Hsueh Tsa Chih (Revue chinoise d'épidémiologie) 1996;17:47-51. 10. Wang S-J, Fuh J-L, Teng EL, et al. A door-to-door survey of Parkinson's disease in a Chinese population in Kinmen. Arch Neurol 1996;53:66-71. 11. Chouza C, Ketzoian C, Caamano JL. Prevalence of Parkinson's disease in a population of Uruguay. Preliminary results. Adv Neurol 1996;69:13-7. 12. Trenkwalder C, Schwarz J, Gebhard J, et al. Starnberg trial on epidemiology of parkinsonism and hypertension in the elderly. Arch Neurol 1995;52:1017-22. 13. de Rijk MC, Tzourio C, Breteler MMB, et al. Prevalence of Parkinsonism and Parkinson's disease in Europe: the EUROPARKINSON collaborative study. J Neurol Neurosurg Psychiatry 1994;62:10-15. 14. Caradoc-Davies TH, Weathers M, Dixon GS, et al. Is the prevalence of Parkinson's disease in New Zealand changing? Acta Neurol Scand 1992;86:40-4. 15. Mayeux R, Marder K, Cote LJ, et al. The frequency of idiopathic Parkinson's disease by age, ethnic group and sex in Northern Manhattan 1988-1993. Am J Epidemiol 1995;142:820-7. 16. Moriwaka F, Tashiro K, Itoh K, et al. Prevalence of Parkinson's disease in Hokkaio, the northernmost island of Japan. Intern Med 1996;35:276-9. 17. Schoenberg BS, Anderson DW, Haerer AF. Prevalence of Parkinson's disease in the biracial population of Copiah County, Mississippi. Neurology 1985;35:841-5. 18. Morens DM, Davis JM, Grandinetti A, et al. Epidemiologic observations on Parkinson's disease: incidence and mortality in a prospective study of middle aged men. Neurology 1996;46:1044-50. 19. Fall PA, Axelson O, Fredriksson M, et al. Age-standardized incidence and prevalence of Parkinson's disease in a Swedish community. J Clin Epidemiol 1996;49:637-41. 20. Nelson LM, Van Sen Eeden SK, Tanner CM, et al. Incidence of idiopathic Parkinson's disease (PD) in a health maintenance organization (HMO): variation by age, gender and race/ethnicity. Neurology 1997;48:A334. 21. Snow B, Wiens M, Hertzman C, Calne D. A community survey of Parkinson's disease. Can Med Assoc J 1989;141:418-22. 22. Svenson LW, Platt GH, Woodhead SE. Geographic variations in the prevalence rates of Parkinson's disease in Alberta. Can J Neurol Sci 1993;20:307-11. 23. Moghal S, Rajput A, D'Arcy C, et al. Prevalence of movement disorders in elderly community residents. Neuroepidemiology 1994;13:175-8. 24. Moghal S, Rajput A, Meleth R, et al. Prevalence of movement disorders in institutionalized elderly. Neuroepidemiology 1995;14:297-300. 25. Ho SC, Woo J, Lee CM. Epidemiologic study of Parkinson's disease in Hong Kong. Neurology 1989;39:1314-8. 26. Mitchell SL, Kiely DK, Kiel DP, et al. The epidemiology, clinical characteristics and natural history of older nursing home residents with a diagnosis of Parkinson's disease. J Am Geriatr Soc 1996;44:394-9. 27. Kurtzke JF, Flaten TP, Murphy FM. Death rates from Parkinson's disease in Norway reflects increased survival. Neurology 1991;41:1665-7. 28. Chio A, Magnani C, Tolardo G, et al. Parkinson's disease mortality in Italy, 1951 through 1987. Arch Neurol 1993;50:149-53. 29. Ben-ShlomoY, Finnan F, Allwright S, et al. The epidemiology of Parkinson's disease in the Republic of Ireland: observations from routine data sources. Irish Med 1993;86:190-4. 30. Imaisumi Y, Kaneko R. Rising mortality from Parkinson's disease in Japan, 1950-1992. Acta Neurol Scand 1995;91:169-76. 31. Ben-Shlomo Y. The epidemiology of Parkinson's disease. Baillières Clin Neurol 1997;6:55-68. 32. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983;219:979-80. 33. Burns RS, Chiueh CC, Markey, SP, et al. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci USA 1983;80:4546-50. 34. Butterfield PG, Valanis BS, Spencer PS, et al. Environmental antecedents of young-onset Parkinson's disease. Neurology 1993;43:1150-8. 35. Morano A, Jimenez-Jimenez FJ, Molina JA, et al. Risk factors for Parkinson's disease: case-control study in the province of Caceres, Spain. Acta Neurol Scand 1994;89:164-70. 36. Rybicki BA, Johnson CC, Uman J, et al. Parkinson's disease mortality and the industrial use of heavy metals in Michigan. Mov Disord 1993;8:87-92. 37. Wang W-Z, Fang X-H, Cheng X-M, et al. A case-control study on environmental risk factors of Parkinson's disease in Tianjin, China. Neuroepidemiology 1993;12:209-18. 38. Marder KS, Logroscino G, Alfaro B, et al. Environmental risk factors for Parkinson's disease in an urban multiethnic community. Neurology 1998;51:279-81. 39. Seidler A, Hellenbrand W, Robra B-P, et al. Possible environmental, occupational and other etiologic factors for Parkinson's disease: a case-control study in Germany. Neurology 1996;46:1328-35. 40. Vieregge P, Friedrick HJ, Rohl A, et al. Multifactorial etiology of idiopathic Parkinson disease: a case-control study [allemand]. Nervenarzt 1994;65:390-5. 41. Koller WC. When does Parkinson's disease begin? Neurology 1992;42:27-31. 42. Semchuk KM, Love EJ, Lee RG. Parkinson's disease and exposure to agricultural work and pesticide chemicals. Neurology 1992;42:1328-35. 43. Schulte PA, Burnett CA, Boenigner MF, et al. Neurodegenerative diseases: occupational occurrence and potential risk factors, 1982 through 1991. Am J Public Health 1996;86:1281-8. 44. Vieregge P, von Maravic V, Friedrick HJ. Lifestyle and dietary factors early and late in Parkinson's disease. Can J Neurol Sci 1992;19:170-3. 45. Semchuk KM, Love EJ, Lee RG. Parkinson's disease: a test of the multifactorial etiologic hypothesis. Neurology 1993;43:1173-81. 46. Hubble JP, Hassanein R, Cao T, et al. Herbicide-pesticide exposure independent of rural living as a risk factor for Parkinson's disease [résumé]. Neurology 1992;42:174. 47. Hertzman C, Wiens M, Snow B, et al. A case-control study of Parkinson's disease in a horticultural region of British Columbia. Mov Disord 1994;9:69-75. 48. Liou HH, Tsai MC, Chen CJ et al. Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology 1997;48:1583-8. 49. De Michele G, Filla A, Volpe G, et al. Environmental and genetic risk factors in Parkinson's disease: a case-control study in southern Italy. Mov Disord 1996;11:17-23. 50. Jimenez-Jimenez FJ, Mateo D, Gimenez-Roldan S. Exposure to well water and pesticides in Parkinson's disease: a case-control study in the Madrid area. Mov Disord 1992;7:149-52. 51. Tanner CM. Occupational and environmental causes of parkinsonism. Occup Med 1992;7:503-13. 52. Gorell JM, Johnson CC, Rybicki BA, et al. Occupational exposures to metals as risk factors for Parkinson's disease. Neurology 1997;48:650-8. 53. Jimenez-Jimenez FJ, Molina JA, Aquilar MV, et al. Serum and urinary manganese levels in patients with Parkinson's disease. Acta Neurol Scand 1995;91:317-20. 54. Chaturvedi S, Ostbye T, Stoessl AJ, et al. Environmental exposures in elderly Canadians with Parkinson's disease. Can J Neurol Sci 1995;22:232-4. 55. Marsden CD. Parkinson's disease in twins. J Neurol Neurosurg Psychiatry 1986;50:105-6. 56. Martyn CN, Osmond C. Parkinson's disease and the environment in early life. J Neurol Sci 1995;132:201-6. 57. Mayeux R, Tang M-X, Marder K, et al. Smoking and Parkinson's disease. Mov Disord 1994;9:207-12. 58. Tzourio C, Rocca WA, Breteler MM, et al. Smoking and Parkinson's disease: an age-dependent risk effect? Neurology 1997;49:1267-72. 59. Hellenbrand W, Seidler A, Robra B-P, et al. Smoking and Parkinson's disease: a case control study in Germany. Int J Epidemiol 1997;26:328-39. 60. Grandinetti A, Morens DM, Reed D, et al. Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson's disease. Am J Epidemiol 1994;139:1129-38. 61. Prasad C, Ikegami H, Shimizu I, et al. Chronic nicotine intake decelerates aging of nigrostriatal dopaminergic neurons. Life Sci 1994;54:1169-84. 62. Riggs JE. Cigarette smoking and Parkinson disease: the illusion of a neuroprotective effect. Clin Neuropharmacol 1992;15:88-9. 63. Cerhan JR, Wallace RB, Folsom AR. Antioxidant intake and risk of Parkinson's in older women [résumé]. Am J Epidemiol 1994;139:S65. 64. Logroscino G, Marder K, Cote L, et al. Dietary lipids and antioxidants in Parkinson's disease: a population-based, case-control study. Ann Neurol 1996;39:89-94. 65. Morens DM, Grandinetti A, Waslien CI, et al. Case-control study for idiopathic Parkinson's disease and dietary vitamin E intake. Neurology 1996;46:1270-4. 66. Gorell JM, Johnson CC, Rybicki BA, et al. A population-based case-control study of nutrient intake in Parkinson's disease. Neurology 1997;48:A298. 67. de Rijk MC, Breteler MMB, den Breeijen JH, et al. Dietary antioxidants and Parkinson disease. The Rotterdam Study. Arch Neurol 1997;54:762-5. 68. Scheider WL, Hershey LA, Vena JE, et al. Dietary antioxidants and other dietary factors in the etiology of Parkinson's disease. Mov Disord 1997;12:190-6. 69. Fernandez-Calle P, Molina JA. Levels of alpha-tocopherol (vitamin E) in Parkinson's disease. Neurology 1992;42:1064-6. 70. King D, Palyfer JR, Roberts NB. Concentration of vitamins A, C and E in elderly patients with Parkinson's disease. Postgrad Med J 1992;68:634-7. 71. Dexter DT, Ward RJ, Wells FR, et al. Alpha-tocopherol levels in brain are not altered in Parkinson's disease. Ann Neurol 1992;32:591-3. 72. Coughlin SS, Pincus JH, Karstaedt P. An international comparison of dietary protein consumption and mortality from Parkinson's disease. J Neurol 1992;239:236-7. 73. Hellenbrand W, Seidler A, Boeing H, et al. Diet and Parkinson's disease I: A possible role for the past intake of specific foods and food groups. Results from a self-administered food frequency questionnaire in a case-control study. Neurology 1996;47:636-43. 74. Jimenez-Jimenez FJ, Mateo D, Gimenez-Roldan S. Premorbid smoking, alcohol consumption and coffee drinking habits in Parkinson's disease: a case-control study. Mov Disord 1992;7:339-44. 75. Zack MM, Tanner CM. Infectious diseases as they relate to the etiology of Parkinson's disease. Dans: Ellenberg JH, Koller WC, Langston JW, réds. Etiology of Parkinson's disease. New York: Marcel Dekker, Inc., 1995:401-23. 76. Hubble JP, Cao T, Kjelstrom JA, et al. Nocardia species as an etiologic agent in Parkinson's disease: serological testing in a case-control study. J Clin Microbiol 1995;33:2768-9. 77. De Pedro-Cuesta J, Petersen IJ, Stawiarz L, et al. High levodopa use in periodically time-clustered, Icelandic birth cohorts. A vestige of parkinsonism etiology? Europarkinson Preparatory Activity Research Group. Acta Neurol Scand 1995;91:79-88. 78. Fazzini E, Fleming J, Fahn S. Cerebrospinal fluid antibodies to coronaviruses in patients with Parkinson's disease. Mov Disord 1992;7:153-8. 79. Altschuler E. Gastric helicobacter pylori infection as a cause of idiopathic Parkinson disease and non-arteric anterior optic ischemic neuropathy. Med Hypotheses 1996;47:413-4. 80. Ward CD, Duvoisin RC, Ince SE, Nutt JD, Eldridge R, Calne DB. Parkinson's disease in 65 pairs of twins and in a set of quadruplets. Neurology 1983;33:815-24. 81. Marttila RJ, Kaprio J, Koskenvuo M, Rinne UK. Parkinson's disease in a nationwide twin cohort. Neurology 1988;38:1217-9. 82. Payami H, Larsen K, Bernard S, et al. Increased risk of Parkinson's disease and siblings of patients. Ann Neurol 1994;36:659-61. 83. Bonifati V, Fabirzio,E, Vanacore N, et al. Familial Parkinson's disease: a clinical genetic analysis. Can J Neurol Sci 1995;22:272-9. 84. Vieregge P. Genetic factors in the etiology of idiopathic Parkinson's disease. J Neural Transm Park Dis Dement Sect 1994;8:1-37. 85. Marder KS, Tang M-X, Mejia H, et al. Risk of Parkinson's disease among first degree relatives: a community based study. Neurology 1996;47:155-60. 86. Uitti RJ, Shinotoh H, Mayward M, et al. "Familial Parkinson's disease"-a case-control study of families. Can J Neurol Sci 1997;24:127-32. 87. Wszolek,ZK, Pfeiffer B, Fulgham JR, et al. Western Nebraska family (family D) with autosomal dominant parkinsonism. Neurology 1995;45:502-5. 88. Golbe LI, Di Iorio G, Sanges G, et al. Clinical genetic analysis of Parkinson's disease in the Contursi kindred. Ann Neurol 1996;43:767-75. 89. Markopoulou K, Wszolek ZK, Pfeiffer RF. A Greek-American family with autosomal dominant levodopa-responsive parkinsonism. Ann Neurol 1995;38:373-8. 90. Bonucelli O, Piccini P, del Dotto,P. Apomorphine test for dopaminergic responsiveness: a dose assessment study. Mov Disord 1993;8:158-64. 91. Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klinische Wochenschrift 1960;38:1236-9. 92. Birkmayer W, Hornykiewicz O. The effect of L-3,4-dihydroxyphenylalanine (= DOPA) on akinesia in Parkinsonism. Wiener Klinische Wochenschrift 1961;73:787-8. 93. Polymeropoulos MH, Higgins JJ, Golbe LI, et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science 1996;274:1197-9. 94. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the a-synclein gene identified in families with Parkinson's disease. Science 1997;276:2045-7. 95. Landi T, Ceroni M, Martignoni E, et al. Gene-environment interaction in Parkinson's disease. The case of CYP2D6 gene polymorphism [survol]. Adv Neurol 1996;69:61-72. 96. Barbeau A, Cloutier T, Roy M. Ecogenetics of Parkinson's disease: 4-hydroxylation of debrisoquine. Lancet 1985;2:1213-6. 97. Steventon MJ, Lledo P, Quinn NP, et al. Degenerative neurological disease and debrisoquine-4-hydroxylation capacity. Med Sci Res 1989;17:163-4. 98. Benitez J, Ladero JM, Jimenez-Jimenez FJ. Oxidative polymorphism of debrisoquine in Parkinson's disease. J Neurol Neurosurg Psychiatry 1990;53:289-92. 99. Meillard NM, Bentue-Ferrer D, Bentue-Ferrer,F, et al. Étude de l'hydroxylation de la débrisoquine dans la maladie de Parkinson. Presse Méd 1990;19:947-9. 100. Gudjonsson,O, Sanz E, Alvan G, et al. Poor hydroxylator phenotypes of debrisoquine and S-mephenytoin are not over represented in a group of Parkinson's disease patients. Br J Clin Pharmacol 1990;30:301-2. 101. Kallio J, Martilla RJ, Rinne UK, et al. Debrisoquine oxydation in Parkinson's disease. Acta Neurol Scand 1991;83:194-7. 102. Steiger MJ, Lledo P, Quinn NP, et al. Debrisoquine hydroxylation in Parkinson's disease. Acta Neurol Scand 1992;86:159-64. 103. Armstrong M, Daly AK, Cholerton S, et al. Mutant debrisoquine hydroxylation genes in Parkinson's disease. Lancet 1992;339:1017. 104. Smith CAD, Gough AC, Leigh PN, et al. Debrisoquine hydroxylase gene polymorphism and susceptibility to Parkinson's disease. Lancet 1992;339:1375. 105. Gasser T, Muller-Myhsok B, Supala A, et al. The CYP2D6B allele is not overrepresented in a population of German patients with idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 1996;61:518-20. 106. Kurth MC, Kurth JH. Variant cytochrome P450 CYP2D6 allellic frequencies in Parkinson's disease. Am J Med Genet 1993;48:166-8. 107. Plante-Bordeneuve V, Davis MB, Maraganore DM, et al. Debrisoquine hydroxylase gene polymorphism in familial Parkinson's disease. J Neurol Neurosurg Psychiatry 1994;57:911-3. 108. Agundez JA, Jimenez-Jimenez FJ, Luengo A, et al. Association between the oxidative polymorphism and early onset of Parkinson's disease. Clin Pharmcol Ther 1995;57:291-8. 109. Diederich N, Hilger C, Goetz CG, et al. Genetic variability of the CYP 2D6 is not a risk factor for sporadic Parkinson's disease. Ann Neurol 1996;40:463-5. 110. Ray-Chadhuri K, Smith C, Gough AC, et al. Debrisoquine hydroxylase gene polymorphism in Parkinson's disease and amylotrophic lateral sclerosis [lettre]. J Neurol Neurosurg Psychiatry 1995;58:109. 111. Kondo I, Kanazawa I. Debrisoquine hydroxylase and Parkinson's disease. Adv Neurol 1993;60:338-42. 112. Sandy MS, Armstrong M, Tanner CM, et al. CYP 2D6 allelic frequencies in young-onset Parkinson's disease. Neurology 1996;47:225-30. 113. Takakubo F, Yamamoto M, Ogawa N, et al. Genetics association between cytochrome P450Ia1 gene and susceptibility to Parkinson's disease. J Neural Transm Gen Sect 1996;103:843-9. 114. Coleman T, Ellis SW, Martin IJ. 1-methyl-2-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is N-demethylated by cytochromes P450 2D6,1A2,3A4-implications for susceptibility to Parkinson's disease. J Pharm Exp Therap 1996;277:685. 115. Bandmann O, Vaughan J, Holmans P, Marsden CD, Wood NW. Association of slow acetylater genotype for N-acetyltransferase 2 with familial Parkinson's disease. Lancet 1997;350:1136-9. 116. Schapira AHV, Cooper JM, Dexter D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1989;1:1269. 117. Schapira AHV, Cooper JM, Dexte, D. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 1990;54:823-7. 118. Mizuno Y, Ohta S, Tanaka M, et al. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem Biophys Res Comm 1989;163:1450-5. 119. Swerlow RH, Parks JK, Miller SW, et al. Origin and functional consequence of the complex I defect in Parkinson's disease. Ann Neurol 1996;40:663-71. 120. Bindoff LA, Birch-Machin MA, Cartildge NEF, et al. Respiratory chain abnormalities in skeletal muscles from patients with Parkinson's disease. J Neurol Sci 1991;104:203-8. 121. Schoffner JM, Watts RL, Juncos JL, et al. Mitochondrial oxidative phosphorylation defects in Parkinson's disease. Ann Neurol 1991;30:332-9. 122. Peterson C, Goldman JE. Alterations in calcium content and biochemical processes in cultured skin fibroblasts from aged and Alzheimer's donors. Proc Natl Acad Sci USA 1986;83:2758-62. 123. Krige D, Carroll MT, Cooper JM, et al. Platelet mitochondrial function in Parkinson's disease. Ann Neurol 1992;32:782-8. 124. Parker WD, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 1989;26:716-23. 125. Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by MPP+, a metabolite of the neurotoxin MPTP. Life Sci 1985;36:2503-8. 126. Wallace DC, Shoffner JM, Trugman JM, et al. Mitochondrial DNA mutations in human degenerative diseases and aging. Biochem Biophys Acta 1995;1271:141-51. 127. Clayton DA. Replication of animal mitochondrial DNA. Cell 1982;28:693-705. 128. Ikebe S, Tanaka,M, Ohno K. Increase of deleted mitochondrial DNA in the striatum in Parkinson's disease and senescence. Biochem Biophys Res Comm 1989;163:1450-5. 129. Mann,VM, Cooper,JM, Schapira AHV. Quantitation of a mitochondrial DNA deletion in Parkinson's disease. FEBS 1992;299:218-22. 130. Sandy MS, Langston JW, Smith MT, et al. PCR analysis of platelet mtDNA: lack of specific changes in Parkinson's disease. Mov Disord 1993;8:74-82. 131. Shan DE, Yeh SI, Wan YC, et al. Absence of 4,977-bp deletion of blood cell mitochondrial DNA in patients with young-onset Parkinson's disease. Acta Neurol Scand 1995;91:149-52. 132. Brown MD, Shoffner JM, Kim YL, et al. Mitochondrial DNA sequence analysis of four Alzheimer's and Parkinson's disease patients. Am J Med Genet 1996;61:283-9. 133. Ikebe S, Tanaka M, Ozawa T. Point mutations of mitochondrial genome in Parkinson's disease. Brain Res Mol Brain Res 1995;28:281-95. 134. Shoffner JM, Brown MD, Torroni A, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 1993;17:171-84. 135. Tanaka M, Kovalenko SA, Gong JS, et al. Accumulation of deletions and point mutations in mitochondrial genome in degenerative diseases. Ann N Y Acad Sci 1996;786:102-11. 136. Zweig RM, Singh A, Cardillo JE, et al. The familial occurrence of Parkinson's disease: lack of evidence for maternal inheritance. Arch Neurol 1992;49:1205-7. 137. Wooten GF, Currie LJ, Bennett JP, et al. Maternal inheritance in Parkinson's disease. Ann Neurol 1997;41:265-8. 138. Bonifati V, Vanacore N, Meco G. Anticipation of onset age in familial Parkinson's disease. Neurology 1994;44:1978. 139. Morrison PJ, Godwin-Austen RB, Raeburn JA. Familial autosomal dominant dopa responsive Parkinson's disease on three living generations showing extreme anticipation and childhood onset. J Med Genet 1996;33:504-6. 140. Carrero-Valenzuela R, Lindbland K, Payami, H, et al. No evidence for association of familial Parkinson's disease with CAG repeat expansion. Neurology 1995;45:1760-3. 141. Rubinsztein DC, Leggo J, Goodburn N, et al. Normal CAG and CCG repeats in the Huntington's disease genes of Parkinson's disease patients. Am J Med Genet 1995;60:109-10. 142. Treves TA, Chandra V, Korczyn AD. Parkinson's and Alzheimer's diseases: epidemiological comparison I. Descriptive aspects. Neuroepidemiology 1993;12:336-44. 143. Treves TA, Chandra V, Korczyn AD. Parkinson's and Alzheimer's diseases: epidemiological comparison 2. Persons at risk. Neuroepidemiology 1993;12:345-9. 144. Strittmatter WJ, Saunders AM, Schmechel,D. Apolipoprotein E: high avidity binding to amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer's disease. Proc Natl Acad Sci USA 1993;90:1977-81. 145. Poirier J, Davignon J, Bouthillier D, et al. Apolipoprotein E polymorphism and Alzheimer's disease. Lancet 1993;342:697-9. 146. van Duijn CM, Knijff PD, Cruts, JM. Apolipoprotein E4 allele in a population based study of early onset Alzheimer's disease. Nature Genetics 1994;7:74-8. 147. Kuusisto J, Koivisto K, Kervinen K. Association of apolipoprotein E phenotypes with late onset Alzheimer's disease: population based study. Br Med J 1994;309:636-8. 148. Arai H, Muramatsu T, Higucki S, et al. Apolipoprotein E gene in Parkinson's disease with or without dementia. Lancet 1994;344:889. 149. Benjamin R, Leake A, Edwardson JA, et al. Apolipoprotein E genes in Lewy bodies and Parkinson's disease [lettre]. Lancet 1994;343:1565. 150. Marder K, Maestre G, Cote L, et al. The apolipoprotein E4 allele in Parkinson's disease with and without dementia. Neurology 1994;44:1330-1. 151. Rubinsztein DC, Hanlon CS, Irving RM, et al. Apo E genotypes in multiple sclerosis, Parkinson's disease, schwannomas and late-onset Alzheimer's disease. Mol Cell Probes 1994;8:519-25. 152. Koller WC, Glatt SL, Hubble JP, et al. Apolipoprotein E genotypes in Parkinson's disease with and without dementia. Ann Neurol 1995;37:242-5. 153. Egensperger R, Bancher C, Kosel S, et al. The apolipoprotein E ,4 allele in Parkinson's disease with Alzheimer lesions. Biochem Biophys Res Comm 1996;224:484-6. 154. Ibarreta D, Gomez-Isla T, Portera-Sanchez A, et al. Apolipoprotein E genotype in Spanish patients of Alzheimer's or Parkinson's disease. J Neurol Sci 1995;134:146-9. 155. Whitehead AS, Bertrandy S, Finnan F, et al. Frequency of the apolipoprotein E epsilon 4 allele in a case-control study of early onset Parkinson's disease. J Neurol Neurosurg Psychiatry 1996;61:347-51. 156. Kurth, MC, Lieberman AN, Kurth JH. Genetic evidence that Alzheimer's and Parkinson's disease affect different populations. Mov Disord 1994;9:30. 157. Bandmann O, Davis MB, Marsden CD, et al. Sequence of the superoxide dismutase 1 (SOD 1) gene in familial Parkinson's disease. J Neurol Neurosurg Psychiatry 1995;59:90-1. 158. Nanko S, Ueki A, Hattori M, et al. No allelic association between Parkinson's disease and dopamine D2, D3, and D4 receptor gene polymorphisms. Am J Med Genet 1994;54:361-4. 159. Plante-Bordeneuve V, Davis MB, Maraganore DM, et al. Tyrosine hydroxylase polymorphism in familial and sporadic Parkinson's disease. Mov Disord 1994;9:337-9. 160. Faucheux BA, Nillesse N, Damier P, et al. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson's disease. Proc Natl Acad Sci USA 1995;92:9603-7. 161. Shen XM, Dryhurst G. Further insights into the influence of L-cysteine on the oxidation chemistry of dopamine: reaction pathways of potential relevance to Parkinson's disease. Chem Res Toxicol 1996;9:751-3. 162. Zhang F, Dryhurst G. Effects of L-cysteine on the oxidation chemistry of dopamine: new reaction pathways of potential relevance to idiopathic Parkinson's disease. J Med Chem 1994;37:1084-98. 163. Gatto EM, Carreras MC, Pargament,GA, et al. Neutrophil function, nitric oxide and blood oxidative stress in Parkinson's disease. Mov Disord 1996;11:261-7. 164. Bockelman R, Wolf G, Ransmayr G, et al. NADPH-diaphorase/nitric oxide synthase containing neurons in normal and Parkinson's disease putamen. J Neural Transm Park Dis Dement Sect 1994;7:115-21. 165. Kunugi H, Nanko S, Ueki A, et al. High and low activity alleles of catechol-O-methyltransferase gene: ethnic difference and possible association with Parkinson's disease. Neurosci Lett 1997;221:202-4. 166. Gasser T, Wszolek ZK, Trofatter J, et al. Genetic linkage studies in autosomal dominant parkinsonism: evaluation of seven candidate genes. Ann Neurol 1994;36:387-96. 167. Chase TN. A gene for Parkinson disease. Arch Neurol 1997;54:1156-7. 168. Singer TP, Castagnoli NJ, Ramsay RR, et al. Biochemical events in the development of parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurochem 1987;49:1-8. 169. Heikkila RE, Kindt MV, Sonsalla PK. Importance of monoamine oxidase A in the bioactivation of neurotoxic analogs of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci USA 1988;85:6172-6. 170. Heikkila RE, Hess A, Duvoisin RC. Dopaminergic neuro-toxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science 1984;224:1451-3. 171. Fahn S. The endogenous toxin hypothesis of the etiology of Parkinson's disease and a pilot trial of high-dosage antioxidants in an attempt to slow the progression of the disease. Ann N Y Acad Sci 1989;570:186-96. 172. Riederer P, Konradi C, Hebenstreit G, et al. Neurochemical perspectives to the function of monoamine oxidase. Acta Neurol Scand Suppl 1989;126:41-5. 173. Cohen G. Monoamine oxidase, hydrogen peroxide, and Parkinson's disease. Adv Neurol 1987;45:119-25. 174. Kurth JH, Kurth MC, Poduslo SE, et al. Association of a monoamine oxidase B allele with Parkinson's disease. Ann Neurol 1993;33:368-72. 175. Hotamisligil GS, Girmen AS, Fink JS, et al. Hereditary variations in monoamine oxidase as a risk factor for Parkinson's disease. Mov Disord 1994;9:305-10. 176. Ho SL, Kapadi AL, Ramsden DB, et al. An allelic association study of monoamine oxidase B in Parkinson's disease. Ann Neurol 1995;37:403-5. 177. Morimoto Y, Murayama N, Kuwano A, et al. Association analysis of a polymorphism of the monoamine oxidase B gene with Parkinson's disease in a Japanese population. Am J Med Genet 1995;60:580-72. 178. Nanko S, Ueki A, Hattori M. No association between Parkinson's disease and monoamine oxidase A and B gene polymorphisms. Neurosci Lett 1996;204:125-7. 179. Costa P, Checkoway H, Levy, D, et al. Association of a polymorphism in intron 13 of the monoamine oxidase B gene wth Parkinson disease. Am J Med Genet 1997;74:154-6. 180. Franklin GM, Checkoway H, Costa-Mallen P, et al. The "protective" effect of smoking in Parkinson's disease is modified by an MAO-B polymorphism. Neurology 1997;48:A334. 181. Duvoisin RC, Sage J. The first symptoms. Dans: Duvoisin RC, Sage J, réds. Parkinson's disease. A guide for patient and family. New York: Lippincott-Raven, 1996:13-20. 182. Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. Neurology 1967;17:427-442. 183. Abrams WB, Beers MH, Berkow R. The Merck manual of geriatrics. 2e éd. Whitehouse Station (NJ):Merck Research Laboratories, 1995. 184. Tanner CM. Early intervention in Parkinson's disease: epidemiologic considerations. Ann Epidemiol 1996;6:438-41. 185. Kishore A, Snow BJ. Drug management of Parkinson's disease. Can Fam Phys 1996;42:946-52. 186. Stacy M, Brownlee HJ. Treatment options for early Parkinson's disease. Am Fam Phys 1996;53:1281-7. 187. Silverstein PM. Moderate Parkinson's disease. Postgrad Med 1996;99:52-68. 188. Calne DB. Treatment of Parkinson's disease. N Engl J Med 1993;329:1021-8. 189. Fahn S. Adverse effects of levodopa. Dans: Olanow CW, Lieberman AN, réds. The scientific basis for the treatment of Parkinson's disease. Carforth (Angleterre) :135 Parthenon, 1992:89-112. 190. Hely MA, Morris JG. Controversies in the treatment of Parkinson's disease. Curr Opin Neurol 1996;9:308-13. 191. Olanow CW. Deprenyl in the treatment of Parkinson's disease: clinical effects and speculations on mechanism of action. J Neural Transm Suppl 1996;48:75-84. 192. Sweeney PJ. Parkinson's disease: managing symptoms and preserving function. Geriatrics 1995;50:24-31. 193. Oertel WH, Quinn NP. Parkinson's disease: drug therapy. Baillières Clin Neurol 1997;6:89-108. 194. Stevenson T. Drug therapy in the management of Parkinson's disease. Br J Nurs 1997;6:144-148, 150. 195. Fazzini E. A comparison of neurosurgical procedures in the treatment of Parkinson's disease. Am Park Dis Assoc Newsletter 1993; Spring:1,4. 196. Obeso JA, Rodriguez MC, Gorospe A, et al. Surgical treatment of Parkinson's disease. Baillières Clin Neurol 1997;6:125-45. 197. Shannon KM, Penn RD, Kroin JS, et al. Stereotactic pallidotomy for the treatment of Parkinson's disease: efficacy and adverse effects at 6 months in 26 patients. Neurology 1998;50:434-8. 198. Tasker RR. Deep brain stimulation is preferable to thalamotomy for tremor suppression. Surg Neurol 1998;49:145-53. 199. Duvoisin RC, Sage J. Dietary considerations. Dans: Duvoisin RC, Sage J, réds. Parkinson's disease. A guide for patient and family. New York: Lippincott-Raven, 1996:105-12. 200. Duvoisin RC, Sage J. Common sense about exercise. Dans: Duvoisin RC, Sage J, réds. Parkinson's disease. A guide for patient and family. New York: Lippincott-Raven, 1996:113-26. 201. Rajput AH, Uitti RJ, Offord KP. Timely levodopa (LD) administration prolongs survival in Parkinson's disease. Parkinsonism and Related Disorders 1997;3:159-65. 202. Ben-Shlomo Y, Marmot MG. Survival and cause of death in a cohort of patients with Parkinsonism: possible clues to aetiology? J Neurol Neurosurg Psychiatry 1995;58:293-9. 203. Ebmeier KP, Calder SA, Crawford JR, et al. Parkinson's disease in Aberdeen: survival after 3.5 years. Acta Neurol Scand 1990;81:294-9. 204. Louis ED, Marder K, Cote L, Tang M, Mayeux R. Mortality from Parkinson disease. Arch Neurol 1997;54:260-4. 205. Bennett DA, Beckett LA, Murray AM, et al. Prevalence of parkinsonian signs and associated mortality in a community population of older people. N Engl J Med 1996;334:71-6. 206. Goetz CG, Stebbins GT. Mortality and hallucinations in nursing home patients with advanced Parkinson's disease. Neurology 1995;45:669-71. 207. Chia LG, Liu LH. Parkinson's disease in Taiwan: an analysis of 215 patients. Neuroepidemiology 1992;11:113-20. 208. Gorell JM, Johnson CC, Rybicki BA. Parkinson's disease and its comorbid disorders: an analysis of Michigan mortality data 1970 to 1990. Neurology 1994;44:1865-8. 209. Nakashima K, Maeda M, Tabata M, et al. Prognosis of Parkinson's disease in Japan. Eur Neurol 1997;38:60-3. 210. Moller H, Mellemkjaer L, McLaughlin JK, et al. Occurrence of different cancers in patients with Parkinson's disease. Br Med J 1995;310:1500-1. 211. Finch EJL, Ramsay R, Katona CLE. Depression and physical illness in the elderly. Clin Geriatr Med 1992;8:275-87. 212. Stern Y, Marder K, Tang M-X, et al. Antecedent clinical features associated with dementia in Parkinson's disease. Neurology 1993;42:1690-2. 213. Starkstein SE, Mayberg HS, Leiguarda R, et al. A prospective longitudinal study of depression, cognitive decline and physical impairments in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 1992;55:377-82. 214. Tandberg E, Larsen JP, Aarsland D, et al. The occurrence of depression in Parkinson's disease. Arch Neurol 1996;53:175-9. 215. Tandberg E, Larsen JP, Aarsland D, et al. Risk factors for depression in Parkinson's disease. Arch Neurol 1997;54:625-30. 216. Berrios GE, Campbell C, Politynska BE. Autonomic failure, depression and anxiety in Parkinson's disease. Br J Psychiatry 1995;16:789-92. 217. Tison F, Dartigues TF, Auriacombe S, et al. Dementia in Parkinson's disease: a population-based study in ambulatory and institutionalized individuals. Neurology 1995;45:705-8. 218. Dooneied G, Mirabello E, Bell K, et al. An estimate of the incidence of depression in idiopathic Parkinson's disease. Arch Neurol 1992;49:305-7. 219. Statistique Canada. Projections démographiques pour le Canada, les provinces et les territoires, 1993-2016. Ottawa, 1994; Cat. 91-520. 220. Marwick C. Research priorities for Parkinson's recommended. JAMA 1995;274:1257-8.
Références des auteures Natalie Kontakos et Julie Stokes, Division des maladies liées au vieillissement, Bureau des maladies cardio-respiratoires et du diabète, Laboratoire de lutte contre la maladie, Direction générale de la protection de la santé, Santé Canada (fonds fournis par la Division du vieillissement et des aînés, Direction de la santé de la population, Direction générale de la promotion et des programmes de santé, Santé Canada) Correspondance : Julie Stokes, Immeuble LLCM, Santé Canada, pré Tunney, Indice de l'adresse : 0602E2, Ottawa (Ontario) K1A 0L2 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Dernière mise à jour : 2002-10-02 | |||