
Public Health Agency of Canada
www.publichealth.gc.ca
Common menu bar links
Institutional links
Diseases & Conditions
Health & Safety
Research & Statistics
Agency Information
Search Box
E-mail this page
Creutzfeldt-Jakob Disease
Quick Links
CJD-Surveillance System
| Newsletters to Families | Newsletters to Physicians |
December 2006
PDF version ![]() (922 KB)
(922 KB)
[How to download PDF documents]
BULLETIN CJD, in addition to all other prion diseases, is provincially reportable in Canada. Physicians must report these diseases to local health authorities, as mandated by provincial legislation. |
|
2006 Highlights |
CJD-SS Progress Report
Established by Health Canada in 1998 as a national surveillance system for Creutzfeldt-Jakob disease, CJD-SS maintains ongoing surveillance for CJD under the recently formed Public Health Agency of Canada (PHAC). As of December 31, 2005, CJD-SS reported 655 patient referrals including 274 cases of definite and probable CJD, confirmed by pathology or diagnosed on a clinical basis. Autopsy confirmed referrals represented 82.1% of our total CJD cases, nationally ranking among the highest worldwide.
CJD-SS applies criteria set by the World Health Organization (WHO) when assigning a possible, probable or definite diagnosis of CJD. As reflected in graph A, case reports to the system have been relatively steady. This is likely attributable to sustained awareness of the disease, continual reporting by physicians, and the availability of 14-3-3 testing provided by the surveillance system. Over 835 CSF specimens have been processed since we began 14-3-3 protein testing in our laboratory in 1998. Through genetic analysis on 41% of patient referrals, CJD-SS has also facilitated the diagnosis of 19 cases of genetic prion disease including 10 cases of Gerstmann-Sträussler-Scheinker syndrome (GSS).
Graph A: Cases & Referrals to CJD-SS by Year of Reporting
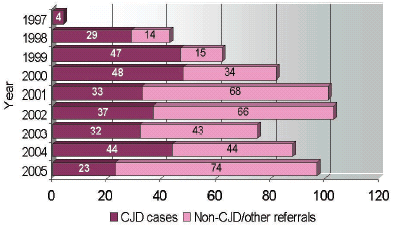
Note: 2005 figures are provisional.
For CJD incidence by year of death, please refer to our statistics.
In addition to laboratory testing, CJD-SS positions field investigators across Canada to complete risk assessment questionnaires by visiting the families of affected individuals. These field investigators also collect diagnostic data by conducting comprehensive medical chart reviews.
From 1999 to 2004, the years for which we have the most complete data thus far, the average incidence of definite/probable CJD is approximately 1.08 per million per year. This rate is consistent with international incidence rates1 of 1 case/ million within the population. The success of CJD-SS surveillance activities is largely credited to ongoing support and cooperation from physicians caring for patients with CJD, and those families affected by this disease.
CJD-SS will continue to use active surveillance methods to identify and investigate all suspected cases in Canada, and assist physicians in diagnosing patients with symptoms of possible or probable CJD. We anticipate notification of future cases primarily through neurologists, pathologists, geriatricians, infectious disease specialists, and infection control practitioners. However, we ask any physician aware of a suspected case of CJD to please contact the Surveillance System using our toll-free number: 1-888-489-2999.
CJD in a Recipient of Tutoplast Dura
Tutoplast Dura is a commercially processed dura mater obtained from human donors and manufactured in Germany. Regulated as a medical device, it was available in Canada between 1982 and 2002 for use in various surgical treatments.
In 2003, as previously reported by various sources, a case of classical CJD was confirmed by autopsy in a patient over the age of 55, who had received a Tutoplast Dura graft in 1992 during neurosurgery. This is the first reported case of CJD allegedly associated with Tutoplast Dura in Canada, although it has not been proven that the patient acquired the disease from the graft. Worldwide, there has been only one previously documented case associated with this particular brand of human dura mater where the graft was considered the most likely source of CJD in the patient2
In April 2002, Health Canada suspended the licence for Tutoplast Dura and monitored a recall of the product. Prior to this action, Tutoplast Dura was the only human cadaveric dura mater product licensed for sale in Canada. Physicians should be aware of the possibility of CJD in recipients of dura mater grafts who develop neurological signs and symptoms. The incubation period can be up to 20 years or more. They are advised to report any suspected cases of CJD in recipients of dura mater grafts to the CJD Surveillance System.
The Role of Human Genetics in Canadian CJD Surveillance
Surveillance efforts have repeatedly shown that a small but important fraction of human prion diseases (ca. 10%) are associated with rare, highly penetrant mutations in the coding region of the prion protein gene (PRNP). Over 20 different point mutations causing amino acid substitutions, and numerous insertions of extra copies of an octapeptide repeat array, have been observed in affected patients.
Phenotypic and pathologic presentation varies from a disease closely resembling classical sporadic CJD, to a distinctive syndrome called Gerstmann-Strãussler- Scheinker syndrome (GSS), fatal familial insomnia (FFI) and other, less easily defined clinical entities. In such challenging cases, discovery of a mutation by molecular analysis of PRNP provides the single most definitive support for a diagnosis of prion disease in the living patient, although the gold standard for diagnosis of prion disease is still reliant on postmortem examination. In addition to these disease-causing mutations, a commonly occurring polymorphism at codon 129, alternately encoding Methionine or Valine in the prion protein, plays a significant role in susceptibility to all known non-genetic forms of prion disease (both infectious and sporadic).
Since its inception, the CJD-SS has provided laboratory services to completely sequence the portion of the PRNP coding region where the above-mentioned mutations and polymorphisms occur, for each suspected CJD patient for whom informed consent is given. In this way, presence and nature of mutations can be conclusively confirmed or disconfirmed, and additional data of significance for surveillance (i.e., codon 129 genotype) are obtained simultaneously. More information on how to draw on these services, as well as supporting technical information, can be obtained by contacting the CJD Surveillance System.
Pathological Examination in CJD
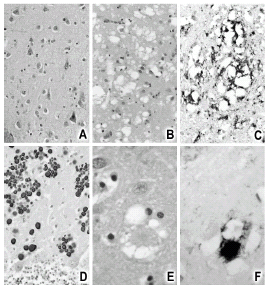
Although a diagnosis of Creutzfeldt- Jakob disease (CJD) can be made quite likely on a clinical basis, pathological examination of brain tissue at autopsy is the golden standard for diagnosis of CJD. The general findings in CJD in the cerebral and cerebellar cortex as well as the basal ganglia are loss of neurons, spongiosis (fig B, compare to A which is normal), also known as vacuolization; and gliosis, the accumulation of astrocytes. Further in immunohistochemical stains the accumulation of a specific protein is seen, the altered prion protein (fig C).
Pathological examination allows variant CJD, Gerstmann-Strãussler-Scheinker syndrome (GSS) and some other rarer genetic forms of CJD to be distinct from this general picture because of special features, like the florid plaque in variant CJD (a round accumulation of prion protein surrounded by a ring of vacuoles seen in fig E, with prion protein immunostain in fig F), and the multicentric plaque in GSS (fig D).
Thus, the autopsy remains an invaluable instrument in detecting CJD and subtyping CJD, even though it occurs only after death.
- Ladogana A et al. Mortality of Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005 May 10; 64 (9)1586-1591.
- Hannah EL, Belay ED, Gambetti P, et al. Creutzfeldt-Jakob disease after receipt of a previously unimplicated brand of dura mater graft. Neurology 2001;56:1080-1083.